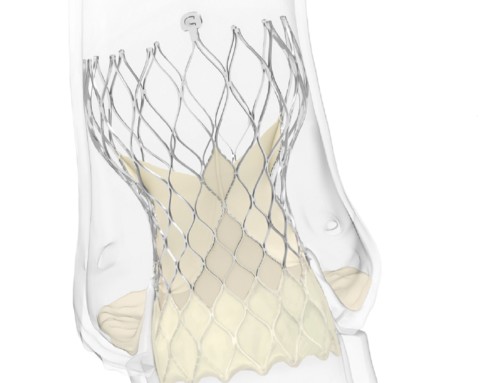
V-Wave has announced that the FDA has just granted the company a Breakthrough Device Designation for its interatrial shunt for heart failure. V-Wave’s minimally invasive, implanted interatrial shunt is being evaluated in a global, randomised, controlled, double-blinded, 500 patient pivotal IDE trial called RELIEVE-HF. The study is enrolling advanced heart failure patients with preserved or reduced left ventricular ejection fraction who remain symptomatic despite the use of guideline directed medical and device therapies.
William T Abraham, V-Wave’s chief medical officer, says: “This FDA Breakthrough Device Designation emphasises the critical and unmet need for novel therapeutic devices for heart failure. More than 6 million people suffer from chronic heart failure in the USA. Heart failure remains a leading cause of acute hospitalization in the Medicare age group. Despite decades of advances in therapy, heart failure patients continue to deteriorate, enduring disabling symptoms, having a poor quality of life, diminished exercise tolerance, and a markedly reduced life expectancy. The V-Wave shunt relieves excessive pressure in the left-side of the heart thereby reducing the build-up of fluid in the lungs, which is known to be the most common reason for heart failure hospitalisations and exercise limitation.”
V-Wave CEO Neal Eigler, comments: “We are thrilled to be able to work even more closely with the FDA to accelerate the introduction of potentially clinically impactful therapies in the USA. This breakthrough designation provides V-Wave with additional options for FDA communication that will facilitate collaboration, as well as a prioritised review of submissions and marketing applications. The potential for early CMS support for this programme, makes our Breakthrough Designation a double-win for heart failure patients who need access to novel therapies as quickly as possible.”