This month, Medtronic has received FDA approval for its Micra AV system and for its Stealth Autoguide system. Also, the FDA gave the company the nod to proceed with an investigational device exemption (IDE) trial to evaluate a pulsed field ablation (PFA) system. Other approvals in January include a FDA green light for a less invasive implantation technique for the HeartMate 3 (Abbott) and the first-ever CE mark for a TMVI system.
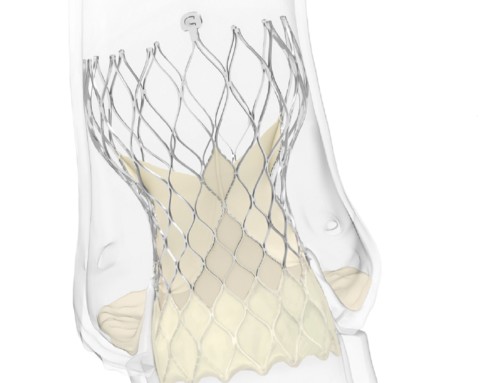
CE mark—Tendyne (Abbott)
The Tenyne mitral valve implantation (TMVI) system (Abbott) has received the CE mark, making Abbott the first company to have such device on a market anywhere in the world. A press release reports that this “life-changing” therapy treats significant mitral regurgitation in patients requiring a heart valve replacement and provides a safe and effective solution for patients who are not candidates for open-heart surgery or transcatheter mitral valve repair.
CE mark extension—Revivent TC less invasive ventricular enhancement therapy (BioVentrix)
BioVentrix has received an extension of its CE mark for the Revivent TC transcatheter ventricular enhancement system to May 2024. The company received its initial CE mark in 2016.
FDA approvals
FDA approval—Micra AV (Medtronic)
Medtronic has received FDA approval of Micra AV, which a press release describes as “the world’s smallest pacemaker with atrioventricular (AV) synchrony”; the pacemaker is indicated for the treatment of patients with AV block.
FDA approval—lateral thoracotomy approach for HeartMate 3 (Abbott)
The FDA has approved a new alternative surgical technique for the HeartMate 3 heart pump (Abbott) that will allow more advanced heart failure patients to avoid open heart surgery. The new, less invasive approach is designed to provide surgeons a choice in surgical method for patients receiving the HeartMate 3 left ventricular assist device (LVAD).
FDA approval—Stealth Autoguide system (Medtronic)
The FDA has cleared the Stealth Autoguide system (Medtronic), which a press release reports is the first cranial robotic platform to integrate with Medtronic’s enabling technology portfolio to create an end-to-end procedural solution.
FDA approval—AI algorithms (EKO)
A press release reports that the FDA has cleared a suite of algorithms that, when combined with Eko’s digital stethoscopes, will enable healthcare providers in the USA to more accurately screen for heart conditions during routine physical exams.
FDA premarket approval—WEB aneurysm embolisation system (Terumo)
MicroVention (Terumo) has received FDA premarket approval (PMA) for the WEB aneurysm embolisation system for the treatment of intracranial wide neck bifurcation aneurysms.
FDA 510(k) clearance—Wattson temporary pacing gudiewire (Teleflex)
Teleflex has received 510(k) clearance from the FDA for its Wattson temporary pacing guidewire. A press release reports that this is the first commercially available bipolar temporary pacing guidewire designed specifically for use during transcatheter aortic valve implantation (TAVI) and balloon aortic valvuloplasty.
FDA 510(k) clearance—OptoWire III (Opsens)
Opsens has received 510(k) clearance from the FDA to market its OptoWire III, a coronary pressure guidewire for physiological measurements such as fractional flow reserve and diastolic pressure ratio (dPR).
FDA 510)(k) clearance—Morph DNA deflectable guide catheter (BioCardia)
BioCardia has announced that the FDA has granted 510(k) clearance for the Morph DNA deflectable guide catheter, which is used to guide the Helix biotherapeutic delivery system during CardiAMP cell therapy delivery in the heart.
FDA 510(k) clearance (expanded indication)—Indigo aspiration system (Penumbra)
Penumbra has announced FDA 510(k) clearance for expanded indication of its Indigo aspiration system. As part of the system, Indigo aspiration catheters and separators are indicated for the removal of fresh, soft emboli and thrombi from vessels of the peripheral arterial and venous systems, and now for the treatment of pulmonary embolism.
FDA trial approval—MitraClip (Abbott)
The FDA has approved a first-of-its-kind clinical trial that will compare the effectiveness of Abbott’s minimally invasive MitraClip device with open heart mitral valve surgical repair in people with primary mitral regurgitation who are eligible for open-heart surgery.
FDA trial approval—Natural Vascular Scaffolding (Alucent Biomedical)
Alucent Biomedical has received FDA approval to proceed with a phase 1 clinical trial to evaluate the safety and efficacy of its revolutionary Natural Vascular Scaffolding (NVS) technology.
FDA Breakthrough Device Designation
FDA Breakthrough Device Designation—Selution SLR AV
MedAlliance has now been granted FDA Breakthrough Device Designation status for its Selution sustained limus release (SLR) sirolimus-eluting balloon for an arteriovenous fistulae indication.
FDA Breakthrough Device Designation—JenaValve TAVI system (JenaValve)
JenaValve Technology has received “Breakthrough Device” designation from the FDA. This designation is for severe aortic regurgitation (AR) and AR-dominant mixed aortic valve disease.
FDA Breakthrough Device Designation—Temporary Spur stent system (Reflow Medical)
Reflow Medical announced that its Temporary Spur stent system—novel retrievable stent technology intended for the treatment of below-the-knee peripheral arterial disease—has been designated for the Breakthrough Devices Program by FDA.